Role of NR4A1-Caveolin-1 Axis in the Orchestration of Mitophagy During Macrophage Senescence
Keywords
Abstract
Background/Aims:
Arteriosclerosis (AS) remains a leading cause of global mortality, with macrophage senescence playing a crucial role in its progression. Senescent macrophages, characterized by oxidative stress and inflammation, exhibit dysregulated mitophagy. However, the underlying mechanisms remain unclear.Methods:
This study explores the role of caveolin-1, a structural protein of caveolae, in NR4A1-mediated mitophagy during oxLDL-induced macrophage senescence. Using gene knockdown and overexpression models, we assessed mitochondrial dysfunction, ROS production, cytokine secretion, and mitophagy activity in murine macrophages.Results:
It revealed that NR4A1 promoted mitochondrial dysfunction and senescence through enhanced ROS production and disrupted mitochondrial potential. Caveolin-1 mediated this effect by facilitating NR4A1-induced mitophagy, as evidenced by colocalization of mitochondria and lysosomes and the activation of Parkin-related pathways. NR4A1 upregulated caveolin-1 expression, forming a signaling axis critical for senescence-associated pro-inflammatory cytokine production.Conclusion:
Overall, our study unraveled The NR4A1-caveolin-1 axis orchestrates mitophagy and inflammation in senescent macrophages, shedding light on AS pathogenesis and suggesting potential therapeutic targets to mitigate macrophage-driven inflammation and oxidative stress.Introduction
Arteriosclerosis (AS)-related cardiovascular and cerebrovascular diseases stand as the foremost cause of global mortality [1]. Despite extensive research, the etiology of AS remains elusive. The interplay of biological aging and cellular aging contributes significantly to the onset and progression of AS [2]. Cellular aging is marked by the cessation of cell proliferation, accompanied by increased cell size, granule accumulation, and metabolic changes. These changes result in the secretion of cytokines that disrupt the microenvironment and drive a cycle of oxidative stress and inflammation, leading to tissue dysfunction [3-5]. Therefore, unraveling the mechanism of cellular aging offers a therapeutic target for AS.
Evidence underscores the presence of senescent cells within AS plaques, characterized by diminished proliferation, growth inhibition, heightened apoptosis, elevated DNA damage, epigenetic modifications, shortened telomeres, and compromised mitochondrial function [6, 7]. Notably, senescent macrophages are discernible in the nascent stages of AS plaque development [8]. Patients with unstable AS plaques exhibit augmented macrophage counts and elevated cholesterol levels [9, 10]. These senescent macrophages express aberrant proteins that potentiate inflammation and oxidative stress reactions, thereby undermining AS plaque stability. Our previous study demonstrated that oxLDL-induced senescent macrophages secrete inflammatory mediators such as matrix metalloproteinases (MMPs) and IL-6, thereby bolstering MMP activity and enhancing cell migratory process [11]. Single-cell RNA sequencing analysis of immune cells from murine atherosclerotic aorta showed that foamy macrophages expressed few inflammatory genes but many lipid-processing genes, suggesting the complex roles of foamy and non-foamy cells in the development of atherosclerosis and accompanied inflammatory responses [12]. Overall, these results implicate the biological functions of macrophages in AS plaque stability modulation.
Mitochondria serve as pivotal drivers of cellular aging. Dysfunction in mitochondria precipitates the excessive accumulation of reactive oxygen species (ROS), thereby exacerbating lipid membrane oxidation, perturbing protein synthesis, inducing DNA damage, and expediting premature cellular senescence [13]. Mitochondrial autophagy, a selective quality-control process, removes damaged or dysfunctional mitochondria. The serine/threonine kinase (PINK1/Parkin) pathway orchestrates selective autophagy by tagging depolarized mitochondria [14]. Studies have found that oxLDL-induced cell senescence robustly enhances mitochondrial autophagic activity, resulting in heightened engulfment of mitochondria by lysosomes, precipitating structural and functional mitochondrial impairment, and subsequently engendering cellular senescence [15, 16]. Nuclear receptor family 4A1 (NR4A1), a nuclear transcription factor, has been shown active in modulating mitochondrial red-ox reactions [17]. in the Our previous study also showed the essential role of NR4A1 in Parkin-mediated mitophagy in aortic endothelial cells [18]. In addition, the biological roles of NR4A1 in monocytes, which is the key driver of atherosclerosis, has also been investigated [19]. It was shown that NR4A1 is expressed in atherosclerotic lesion macrophages and is critical in modulating lipid loading and inflammatory responses [20], deletion of NR4A1 in macrophages towards an inflammatory phenotype and increases atherosclerosis [21]. However, clinical study revealed a positive correlation of NR4A1 expression in PBMCs and level of pro-inflammatory cytokines in type 2 diabetes patients, suggesting complex roles of NR4A1 in regulating the activity of immune cells [22]. Above all, the precise regulatory relationship between NR4A1 and oxLDL-induced mitophagy in senescent macrophages remains unclear.
Caveolae are microdomains on cell membrane specialized for molecule transportation, including mitogen-activated protein kinase (MAPK) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [23]. Caveolin-1, the structural protein of caveolae, assumes to have the additional role as regulatory element for numerous signaling pathways [24]. Caveolin-1’s nexus with cellular aging has been subject to divergent observations [25]. For instance, while inhibition of caveolin-1 attenuates cellular aging in hydrogen peroxide-induced NIH-3T3 cells [26], its deficiency in human fibroblasts has been correlated with mitochondrial dysfunction and accelerated cellular aging [27]. Additional evidence showed that caveolin-1 is located both on the cell membrane and mitochondria, underpinning its intricate interplay with cellular autophagy [28, 29]. Despite this, the precise role of caveolin-1 in regulating mitophagy in the context of senescent macrophages is still not fully understood. Previous studies suggest it may participate in mitochondrial dynamics and stress response pathways, potentially linking it to NR4A1-regulated mitophagy [30].. Overall, the involvement of caveolin-1 in NR4A1 mediated mitophagy in senescent macrophages has yet to be defined.
In this study, we unveiled the biological roles of NR4A1 and caveolin-1 in regulating mitophagy during oxLDL-induced macrophage senescence. Specifically, NR4A1 is involved in oxLDL induced mitophagy via caveolin-1. The expression of caveolin-1 under stimulation is regulated by NR4A1. NR4A1-caveolin-1 signaling axis is essential for the expression of pro-inflammatory effectors in senescent macrophage. Overall, these findings offer novel insights into the regulatory orchestration of mitophagy during macrophage senescence.
Materials and Methods
Statement of Ethics
Human subjects or animals are not invovled in this study.
Key reagents
For cell culture and stimulation, Oxidized Low-Density Lipoprotein (oxLDL) (20605ES10) was purchased from
Yeasen Biotechnology Co., Ltd (Shanghai. CN) for the induction of cell senescence; lipofectamine 2000
(11668027) was purchased from ThermoFisher (USA) for siRNA and DNA transfection. For the induction of
mouse bone marrow derived macrophage (BMDM), mouse m-CSF recombinant protein (PMC2044), and red blood cell
(RBC) lysis buffer (00-4333-57) were was purchased from ThermoFisher (USA).
For RNA extraction and subsequent real-time PCR analysis, MolPure® cell RNA kit (19231ES50), Hieff
UNICON® advanced qPCR SYBR master mix (11185ES08), and Hifair® AdvanceFast One-step RT-gDNA
digestion SuperMix for qPCR (11151ES10) were purchased from Yeasen Biotechnology Co., Ltd (Shanghai. CN);
for Elisa assays, mouse IL-6 ELISA Kit (98027ES96), mouse IL-1β ELISA Kit (98024ES96), and mouse
TNF-α ELISA Kit (98029ES96) were purchased from Yeasen Biotechnology Co., Ltd (Shanghai. CN).
For the analysis of mitophagy via fluorescent microscopy, Lyso-Tracker Red (C1046) and Mito-Tracker Green
(C1048) were purchased from Beyotime Biotech. Inc. (CN). For mitochondria function and related cell
assays, mitochondrial membrane potential assay kit with TMRE (C2001S), the reactive oxygen species assay
kit (S0033M), enhanced mitochondrial membrane potential assay kit with JC-1 (C2003S), senescence
b-galactosidase staining Kit (C0602), and cell mitochondria isolation kit (C3601) were purchased from
Beyotime Biotech. Inc. (CN).
For western blot analysis, HRP Labeled Goat Anti-Mouse IgG (H+L) (A0216), GAPDH mouse monoclonal antibody
(AF0006), Bcl rabbit polyclonal antibody (AF6285), HRP-Labeled goat anti-rabbit IgG (H+L) (A0208),
cytochrome C antibody (AC909), BAX antibody (AB026), ATG-5 rabbit monoclonal antibody (AF2269), Caveolin-1
mouse monoclonal antibody (AF0087) were purchased from Beyotime Biotech. Inc. (CN), phospho-Parkin (Ser65)
antibody (36866), LC3A/B Antibody (4108) and cleaved caspase-9 (Asp315) antibody (9595S) were purchased
from Cell Signaling Technology. Inc. (USA).
Cell culture
Raw264.7 cell was purchased from the American Type Culture Collection (ATCC) (Rockville, MD, USA) and
cultured in lab within 8 passages. RPMI-1640 medium supplemented with 10% fetal bovine serum, 1%
penicillin &streptomycin, and 25mM HEPES buffer (Complete Culture Medium or CCM) was used cell
culture. Mouse bone marrow derive macrophages (BMDMs) were obtained by isolation of bone marrow cells from
6 weeks old C57BL/6 mice and differentiated via L cell media (10%) conditioned CCM. To induce cellular
senescence in vitro , cells were cultured with 100 μM oxidized low-density lipoprotein for 24
h [11, 18].
For the overexpression of NR4A1, C-FLAG tagged mouse NR4A1 (pCMV3-NR4A1-FLAG: MG53939-CF, Sinobiological,
CN) or empty vector were transiently transfected into cells via lipofectamine 2000 according to the
manufacturer’s protocols. 24hr after transfection, the overexpressing efficiency was verified via
real-time PCR, cells were used for stimulation and downstream analysis.
For small RNA interference-based gene knock-down, 5′-GCCGGUGACGUGCAACAAUUU-3′ was used for
NR4A1 knock down, 5′-GUGACUGAGAAGCAAGUGUAU-3′ was used for mouse caveolin-1 knockdown, and
5′-GAGCGAGAAGCAAGUGUACGA was used for human caveolin-1 knockdown. All RNA sequences were designed
via siRNA Wizard Software 3.1 (Invivogen. Inc). The siRNA transfection was performed using lipofectamine
2000 transfection reagent according to the manufacturer’s protocol [31]. The knockdown efficiency of
caveolin-1 was verified via real-time PCR.
Induction of bone marrow derived macrophage (BMDM)
As previously described [32], For the induction of BMDM, 5-6 C57BL/6 mouse were euthanized by appropriate
method approved by ICUAC. Mouse bodies were soaked with 70% ethanol, skin was removed to expose lower
limb. Major muscles near the base of the lower limb were cut to expose the hip joint, femurs were cut near
the base, remining major muscles around tibia were removed. Cut at knee joint, remove major muscles around
tibia, and remove fibula to get femur for subsequent performance. For the isolation of bone marrow cells,
remove the epiphyses of femur that bone marrow can be accessed from the ends with a 23G needle. Flush the
bone marrow into a 50 mL tube on ice by slowly injecting approximately 2–3 mL PBS per bone. The bone
marrow then come out from the other end. Centrifuge bone marrow suspension at 200 × g, 5 min at
4°C, aspirate PBS.
For the differentiation into bone marrow-derived macrophages, resuspend bone marrow (1 ×
107 live cells per mouse) in 1 mL culture medium (pre-warmed DMEM, supplemented with 10% FBS,
1% Penicillin streptomycin, 10% L cell media), and filter through a 100 μm cell strainer. Pipette
suspension (1 × 107 live cells) into total volume of culture medium. Add bone marrow
culture medium to wells (1 mL/well for 12 well plate). Culture at 37°C 5% CO2.Add half
volume of culture medium on day 4 (500 μL/well for 12 well plate). BMDMs were ready for in vitro
stimulation and subsequent analysis at day 6-7.
ROS production analysis
ROS production was measured by DCFH-D staining. According to the manufacturer’s protocol, 96-well
plate seeded samples were washed three times with cold-PBS and then stained with serum-free medium diluted
DCFH-D fluorescent probe (final concentration 10mM) and incubated at 37°C in the dark for 20-30min.
Later, samples were washed 3 times with serum free medium and the fluorescent signal at 525nm was detected
via microplate reader. For flow cytometry analysis, cells were detached, stained with DCFH-D and washed as
above, fluorescent intensity of ROS was then monitored via flow cytometer.
mPTP opening analysis
For mPTP opening assay, tetramethylrhodamine ethyl ester (TMRE) fluorescence was used according to the
manufacturer’s protocol. Similar to ROS staining & plate reading analysis described above, the
fluorescence intensity of TMRE at 575nm was recorded for mPTP opening rate analysis.
Mitochondrial potential analysis
JC-1 staining was used to monitor mitochondrial potential. According to the manufacturer’s protocol,
96-well plate seeded samples were washed three times with cold-PBS and then 10 mg/ml of JC-1 were added
into the medium for approximately 10 min at 37 °C in the dark [33, 34]. Subsequently, cold-PBS was
used to wash the samples three times at room temperature. The mitochondrial potential was analyzed by the
measurement of fluorescent signals at 529nm and 585nm via microplate reader.
Immunofluorescent microscopy
Localization between lysosome and mitochondria were monitored by co-staining of Lyso-Tracker Red
(Excitation wavelength: 577nm; Emission wavelength: 590nm) and Mito-Tracker Green (Excitation wavelength:
490nm; Emission wavelength: 516nm). According to the manufacturer’s protocol, samples were stained
with Lyso-Tracker Red (final concentration 50nM) and Mito-Tracker Green (100nM) at 37°C in the dark
for 30min. Samples were then washed with warmed PBS before monitoring. Leica SP8 LSCM confocal microscope
was used for image recording and Image J software 1.53t (NIH, USA) was used for subsequent analysis.
β-Galactosidase assay
b-Galactosidase staining was performed according to the manufacturer’s protocol. Briefly, samples
were washed 3 times with PBS and fixed with 1ml b-Galactosidase fix solution at room temperature for
15min. Later, samples were washed 3 times with PBS and stained with b-Galactosidase staining mixture at
37°C overnight and monitored under microscope.
Mitochondria isolation
As previously described [35], mitochondria were isolated via the mitochondria isolation kit. Briefly, the
cells were minced and suspended in Reagent A on ice for 10 min and then homogenized with a glass dounce
homogenizer. Then, the lysates were centrifuged at 800 × g for 10 min at 4 °C. The supernatant
was centrifuged at 12000g for 20 min at 4 °C, and the remaining pellet was resuspended in Reagent C
supplemented with phosphatase/protease inhibitor for further experiments.
Western blot
Samples were washed three times with PBS. Samples were then lysed in RIPA buffer to extract proteins and
were then centrifuged at 12000rpm for 10 min at 4 °C. The supernatants were then collected and
subjected to immunoblotting analysis. All primary antibodies used in western blot analysis were specified
in the Reagent section.
Real time PCR
For real-time PCR analysis, samples were washed three times with PBS and mRNA was extracted via the RNA
extraction kit according to the manufacturer’s protocol. The concentration of mRNA was measured and
cDNA was synthesized via reverse transcription PCR according to the manufacturer’s protocol. qPCR
SYBR master mix was used for real time PCR reactions and was performed under CFX Real-Time PCR System
(BioRad, USA). For real-time PCR primers, 5’- GGTGACTGAGAAGCAAGTGTAT-3’ and 5’-
AGGAAGGAGAGAATGGCAAAG-3’ were used for mouse caveolin-1; 5’- GTCAGTGGTCAGTGTGATTGT-3’
and 5’-GTAGTGAAGGCAGAGGTGAAAG-3’ were used for mouse NR4A1. All RNA sequences were designed
via IDT (Integrated DNA Technologies, USA).
Elisa
The concentration of mouse IL-6, TNF-a and IL-1b in cell supernatants were detected via commercialized
Elisa kit following the manufacturer’s protocols. Briefly, samples were incubated in pre-coated
96-well Elisa plate at room temperature for 2hrs, then washed 3 times with PBST buffer. 100ml diluted
detection antibodies, HRP-conjugated antibody and TMB solution were sequentially added for incubation and
interval with 3 times of washing. At last, 50 ml of stop solution was added into each well before plate
reading at 450nm.
Statistical analysis
All data in the present study were presented as the mean ± standard deviation (Standard Error of the
Mean) of three replicates. Difference between two groups were analyzed by t-test. Difference among more
than 2 groups were analyzed by one-way ANOVA followed by post hoc test. Statistical significance threshold
was set as follows: P < 0.05, *; P < 0.01, **; P < 0.001, ***; P < 0.0001, ****.
Results
NR4A1 promotes mitochondria dysfunction during macrophage senescence
To investigate the biological role of NR4A1 in regulating mitochondria functions during macrophage
senescence, we specifically knocked down the expression of NR4A1 in murine macrophage Raw264.7, the
knockdown efficiency was verified via RT-PCR (Fig. 1a) and the activity of mitochondria were tested with
or without oxLDL stimulation. As shown in Fig. 1b, oxLDL treatment significantly reduced mitochondrial
membrane potential (representative indicator for mitochondria function) in wild type cells, while it was
restored by NR4A1 knock down (Fig. 1b). To verify this, we tested the mPTP opening rate, which is another
indicator for mitochondria activity, in different groups. The results revealed that the mPTP opening rate
was remarkably increased in wild type cells upon treatment, while it was substantially suppressed in NR4A1
knock down group (Fig. 1c). In supporting this, ROS production of various groups were also accessed via
both flow cytometry (Fig. 1d) and plate reading assay (Fig. 1e). As showed, NR4A1 deficiency efficiently
inhibited oxLDL induced ROS production. To recapitulate our observations on primary cells, mouse bone
marrow derived macrophages (BMDM) were generated for analysis (Fig. 1f). It showed that NR4A1 knockdown in
BMDM restores the biological functions of mitochondria in response to oxLDL stimulation, as revealed by
membrane potential staining (Fig. 1g). mPTP opening rate (Fig. 1h) and ROS production (Fig. 1i).
To investigate the underlying mechanism of NR4A1 regulated mitochondria dysfunction in response to oxLDL,
the levels of critical proteins involved in the process were also detected. It showed that oxLDL treatment
induced the elevation of cleaved caspase 9 and BAX, the reduction of Bcl-2, as well as the cytosol leakage
of cytochrome C in wild type cells, indicating the dysfunction of mitochondria. However, these effects
were substantially attenuated in NR4A1-deficient cells. (Fig. 2a). To further clarify the regulating
activity of NR4A1 in mitophagy, the mitochondria and lysosome of various groups of cells were stained and
monitored under confocal microscope. It revealed that oxLDL treatment induces the colocalization between
mitochondria and lysosome in wild type group, indicating the presence of mitophagy, while it was
diminished in NR4A1 knock down group (Fig. 2b-c). Overall, these results highlighted the potential role of
NR4A1 in promoting oxLDL induced macrophage senescence.
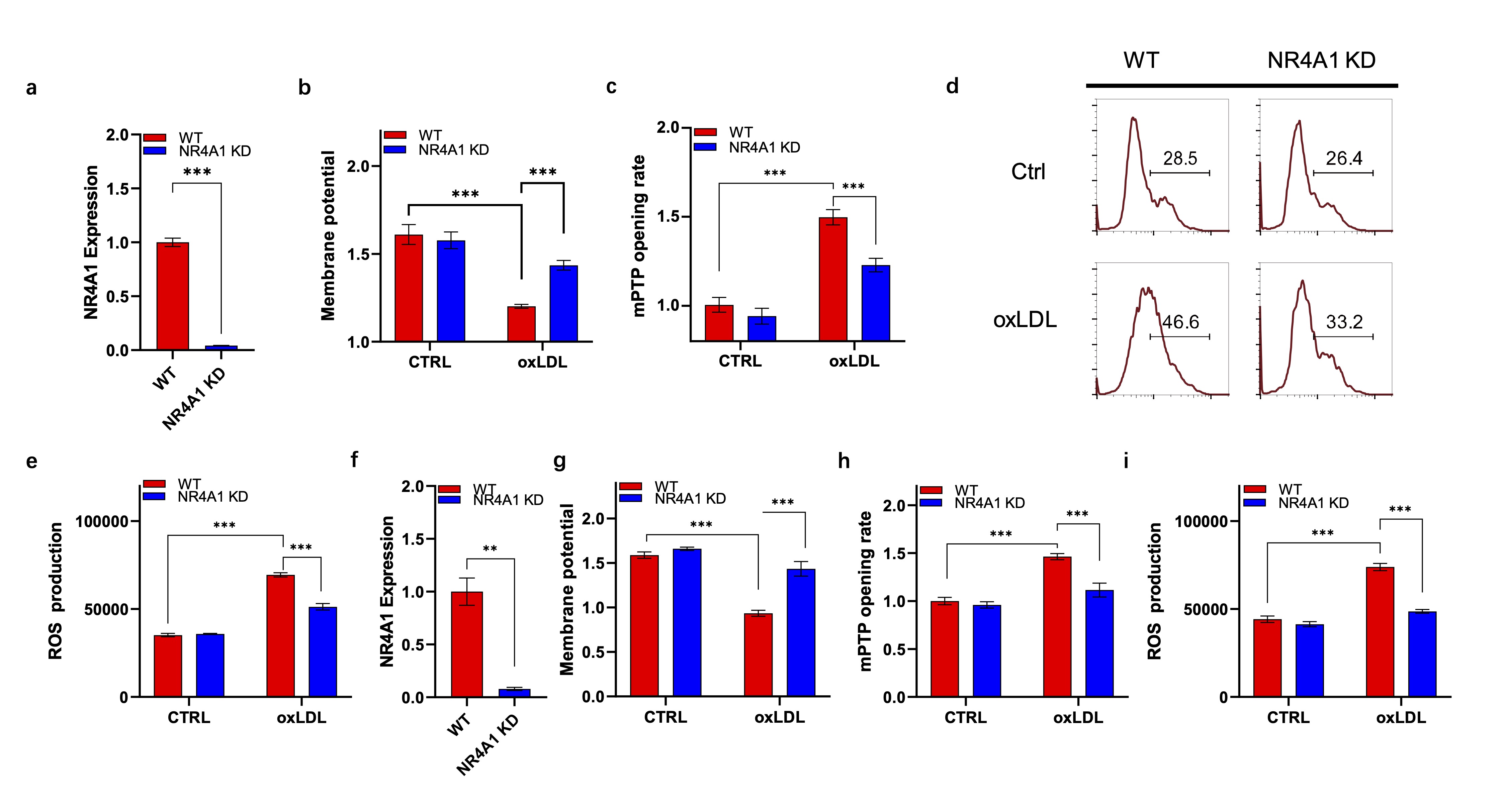
Fig. 1: NR4A1 promotes oxLDL induced macrophage senescence. a). RT-PCR verification of NR4A1 expression in different Raw264.7 cell lines. b) JC-1 staining based membrane potential analysis of wild type and NR4A1 knock down Raw264.7 cells with or without oxLDL stimulation. n=3 (technical repeats). c). mPTP opening analysis of wild type and NR4A1 knocking-down Raw264.7 cells with or without oxLDL stimulation. n=3. d-e). ROS production analysis of wild type and NR4A1 knock down Raw264.7 cells with or without oxLDL stimulation via flow cytometry (d) and plate reading (e). n=3. f). RT-PCR verification of NR4A1 expression in BMDM. g) JC-1 staining based membrane potential analysis of wild type and NR4A1 knocking-down BMDM with or without oxLDL stimulation. n=3 (technical repeats). h). mPTP opening analysis of wild type and NR4A1 knocking-down BMDM with or without oxLDL stimulation. n=3. i) ROS production analysis of wild type and NR4A1 knocking-down BMDM with or without oxLDL stimulation via plate reading analysis. WT: wild type; NR4A1 KD: NR4A1 knock down; CTRL: control.
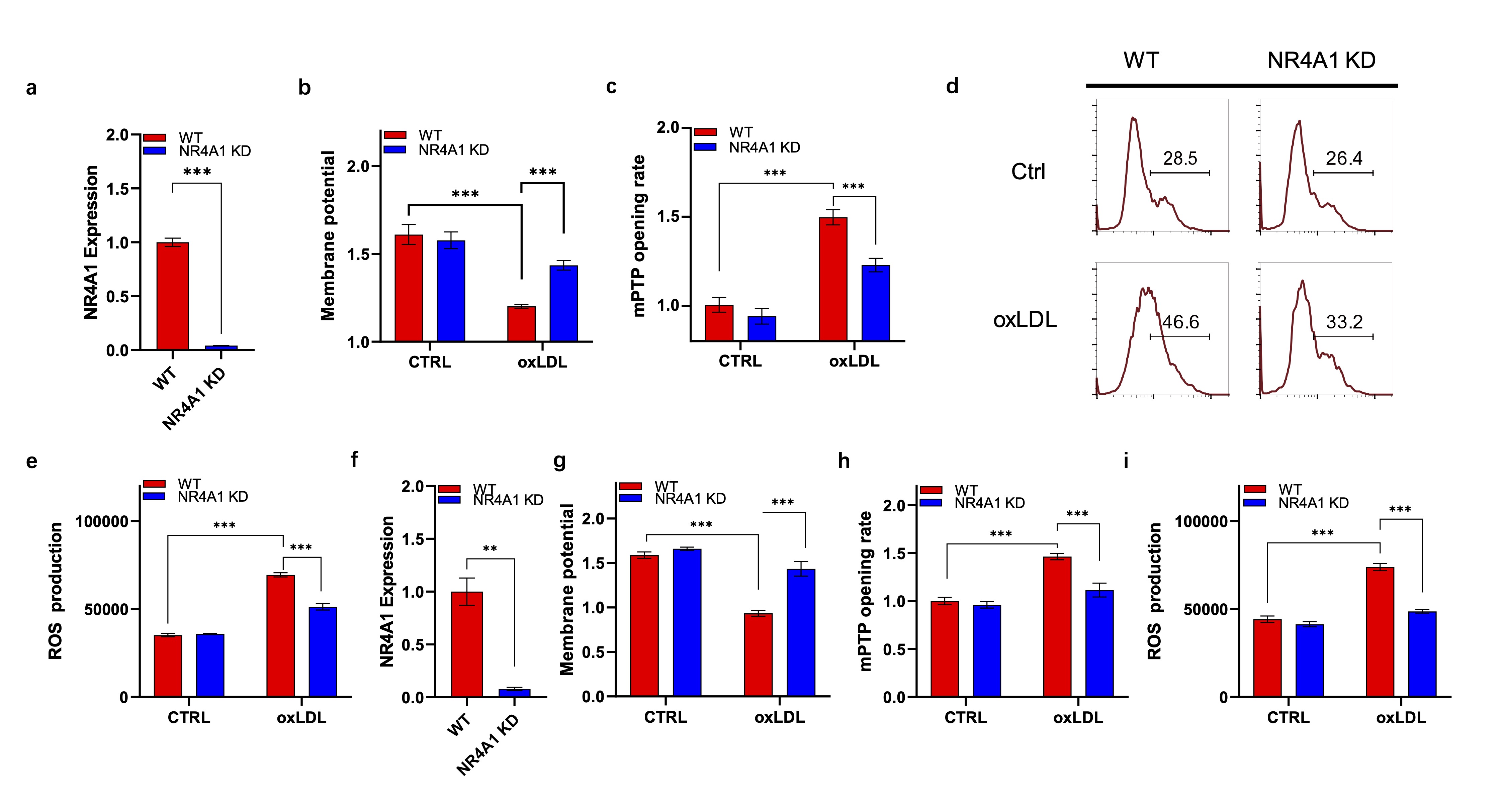
Fig. 2: NR4A1 contributes to mitochondria dysfunction during macrophage senescence. a). Western blot analysis of key proteins in cell death. b-c). Confocal images (b) and colocalization rate analysis (c) between mitochondria (green) and lysosome (red) of wild type and NR4A1 knock down Raw264.7 cells with or without oxLDL stimulation. n=3.
Caveolin-1 mediates NR4A1 induced mitochondria dysfunction
To further unravel the role of caveolin-1 in NR4A1 mediated mitochondria dysfunction, we performed
mitochondrial membrane potential analysis as described above. The knockdown efficiency of caveolin-1 and
NR4A1 was verified by RT-PCR (Fig. 3a-b). It showed that NR4A1 overexpression significantly promoted the
reduction of mitochondrial membrane potential under oxLDL treatment, while it was blocked by the knock
down of caveolin-1 (Fig. 3c). To verify this, we tested ROS production of various groups with or without
oxLDL stimulation. As showed in Fig. 2b, caveolin-1 deficiency significantly reduced the production of
ROS, which promoted by NR4A1 overexpression (Fig. 3d). Western blot analysis revealed that in the presence
of oxLDL, NR4A1 overexpression promoted mitochondrial originated cell death, characterized by cytochrome C
leakage, caspase 9 cleavage, elevation of BAX and reduction of Bcl-2, while it was efficiently restored by
the knock down of caveolin-1 (Fig. 3e). β-galactosidase staining analysis was used to investigate the
role of NR4A1-caveolin-1 axis in oxLDL induced cell senescence. As expected, NR4A1 overexpression
increased oxLDL induced senescence while it was inhibited by caveolin-1 knocking down (Fig. 3f).
Collectively, these results revealed the biological role of caveolin-1 in mediating NR41 transduced
mitochondria dysfunction under oxLDL.
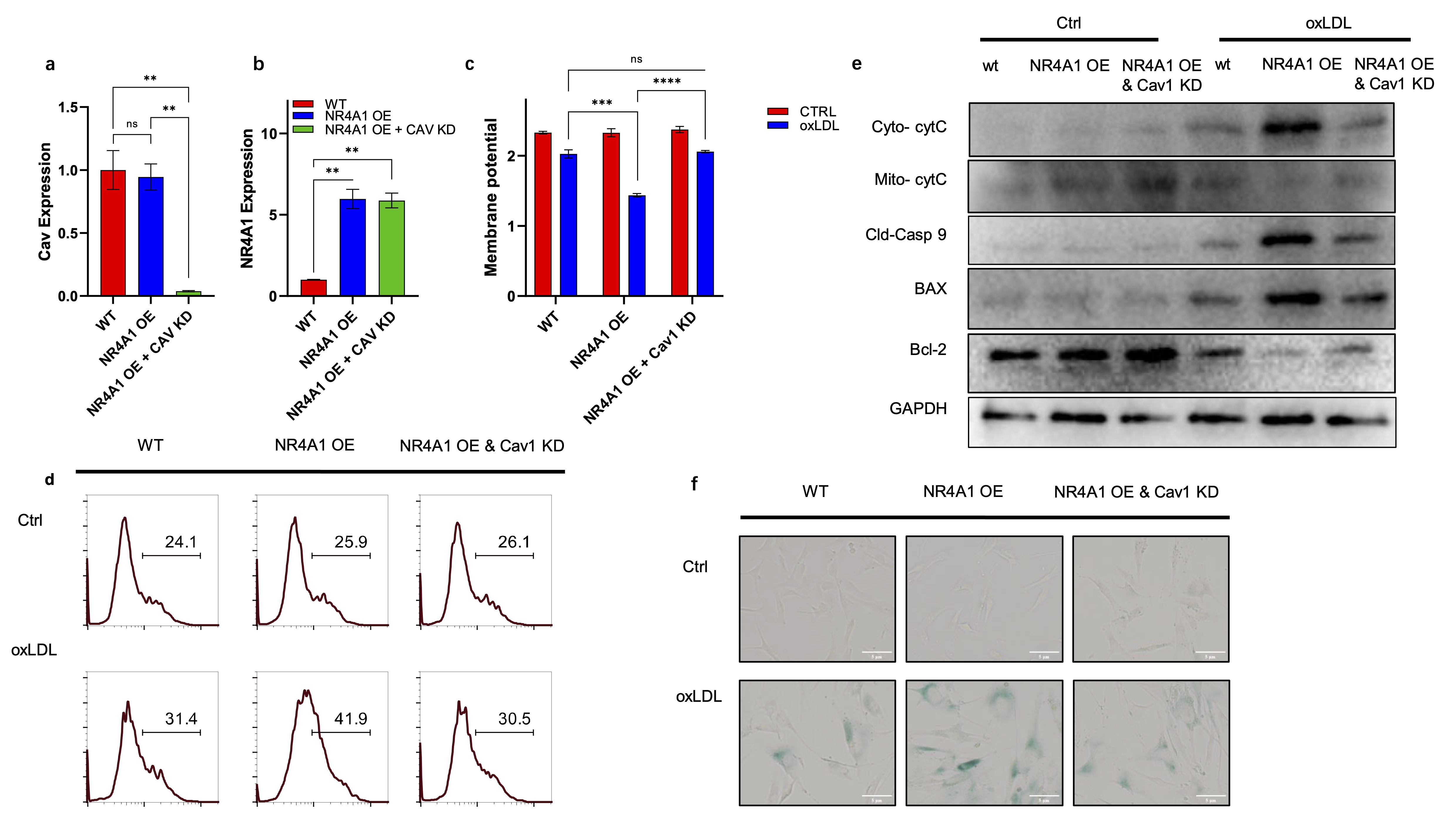
Fig. 3: Caveolin-1 mediates NR4A1 induced mitochondria dysfunction. a). RT-PCR verification of Caveolin-1 expression in different Raw264.7 cell lines. n=3. b). RT-PCR verification of NR4A1 expression in different Raw264.7 cell lines. n=3. c). JC-1 staining based membrane potential analysis of wild type, NR4A1 overexpression, and NR4A1 overexpression plus caveolin-1 knock down Raw264.7 cells with or without oxLDL stimulation. n=3. d). ROS production analysis of wild type, NR4A1 overexpression, and NR4A1 overexpression plus caveolin-1 knock down Raw264.7 cells with or without oxLDL stimulation via flow cytometry. e). Western blot analysis of key proteins in cell death. f). β-Galactosidase staining images of wild type, NR4A1 overexpression, and NR4A1 overexpression plus caveolin-1 knock down Raw264.7 cells with or without oxLDL stimulation. NR4A1 OE: NR4A1 overexpression; NR4A1 OE & Cav1 KD: NR4A1 overexpression plus caveolin-1 knock down.
NR4A1 upregulates caveolin-1 expression during macrophage senescence
Given the synergistic roles of NR4A1 and caveolin-1 in regulating mitochondrial dysfunction, we next
examined whether NR4A1 directly regulates caveolin-1 expression. Real-time PCR analysis showed that oxLDL
stimulation significantly elevated caveolin-1 mRNA levels, while NR4A1 knockdown markedly suppressed this
induction (Fig. 4a). Consistent with this, western blot analysis confirmed reduced caveolin-1 protein
levels in NR4A1-deficient cells under oxLDL treatment (Fig. 4b). These findings were further validated in
primary BMDMs (Fig. 4c-d), indicating that NR4A1 acts upstream of caveolin-1 during macrophage senescence.
These data suggest a regulatory axis where NR4A1 modulates caveolin-1 transcription, possibly through
direct promoter interaction, contributing to the mitophagic and inflammatory phenotype of senescent
macrophages.
Fig. 4: NR4A1 upregulate Caveolin-1 expression during macrophage senescence induction. a). Real-time PCR analysis of mouse caveolin-1 gene in wild type and NR4A1 knocking-down Raw264.7 cells with or without oxLDL stimulation. n=3. b). Western blot analysis of caveolin-1 expressing level in wild type and NR4A1 knock down Raw264.7 cells with or without oxLDL stimulation. n=3.c). Real-time PCR analysis of caveolin-1 expressing levels in wild type and NR4A1 knocking-down BMDM with or without oxLDL stimulation. d). Western blot analysis of caveolin-1 levels in wild type and NR4A1 knocking-down BMDM with or without oxLDL stimulation.
Caveolin-1 promotes NR4A1 mediated mitophagy during macrophage senescence
Previous studies have revealed the essential role of NR4A1 in Parkin related mitophagy [18, 36]. Here we
investigated the involvement of caveolin-1 in mitophagy induction in the downstream of NR4A1. Western blot
analysis showed that NR4A1 overexpression increased the level of phosphorylated Parkin (P-Parkin),
Mitochondrial-retained LC3II, ATG-5, indicating the positive regulating activity of NR4A1 in Parkin
related mitophagy (Fig. 5a). However, the effect of NR4A1 overexpression was blocked by the knock down
Caveolin-1 (Fig. 5a).
To further investigate the activity of caveolin-1 in regulating NR4A1 mediated mitophagy, mitochondria and
lysosome were stained and monitored under confocal microscope. It revealed that NR4A1 overexpression
promoted the colocalization between mitochondria and lysosome under oxLDL stimulation, while it was
inhibited in caveolin-1 knock down group (Fig. 5b). Colocalization analysis showed that the colocalizing
rate between mitochondria and lysosome was significantly reduced by the knock down of caveolin-1 (Fig.
5c). Collectively, these data demonstrated that caveolin-1 is critical in promoting NR4A1 mediated
mitophagy during the senescence of macrophage.
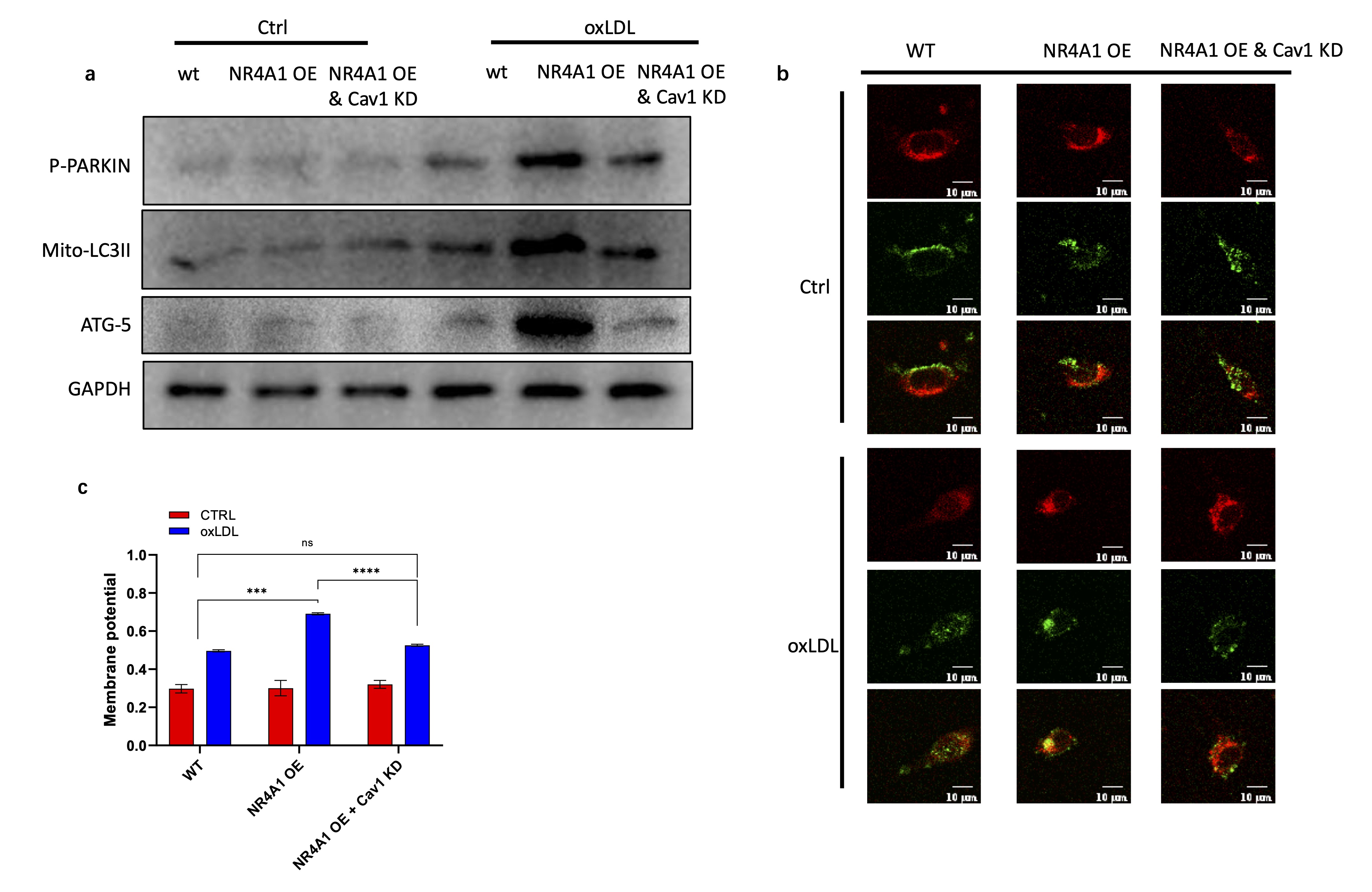
Fig. 5: Caveolin-1 promotes NR4A1 induced mitophagy during macrophage senescence. a). Western blot analysis of key proteins in mitophagy activation. b-c). Confocal images (b) and colocalization rate analysis (c) between mitochondria (green) and lysosome (red) of wild type, NR4A1 overexpression, and NR4A1 overexpression plus caveolin-1 knock down Raw264.7 cells with or without oxLDL stimulation. n=3.
NR4A1-Caveolin-1 signaling axis promotes inflammatory cytokine expression in senescent macrophage
As reported previously, senescent cells produce proinflammatory factors to recruit immune cells and
promote inflammation [37, 38]. To investigate the activity of NR4A1 and caveolin-1 in regulating
inflammation of senescent macrophages, we detected the extracellular level of proinflammatory cytokines
TNF-a, IL-6 and IL-1b of various groups of Raw264.7 cells with or without oxLDL treatment. As revealed,
oxLDL stimulation modestly increased the secretion of the above cytokines, which were significantly
enhanced by the overexpression of NR4A1. However, caveolin-1 knockdown efficiently reduced the
proinflammatory effect of overexpressed NR4A1 (Fig. 6a-c). Additionally, we detected the expression of
Toll-like receptor 4 (TLR4), another proinflammatory receptor on innate immune cells. It showed the
similar pattern as in Fig. 5a to c (Fig. 6d). Overall, these data indicate that the NR4A1-caveolin-1
signaling axis is essential in the onset of inflammation in senescent macrophages.
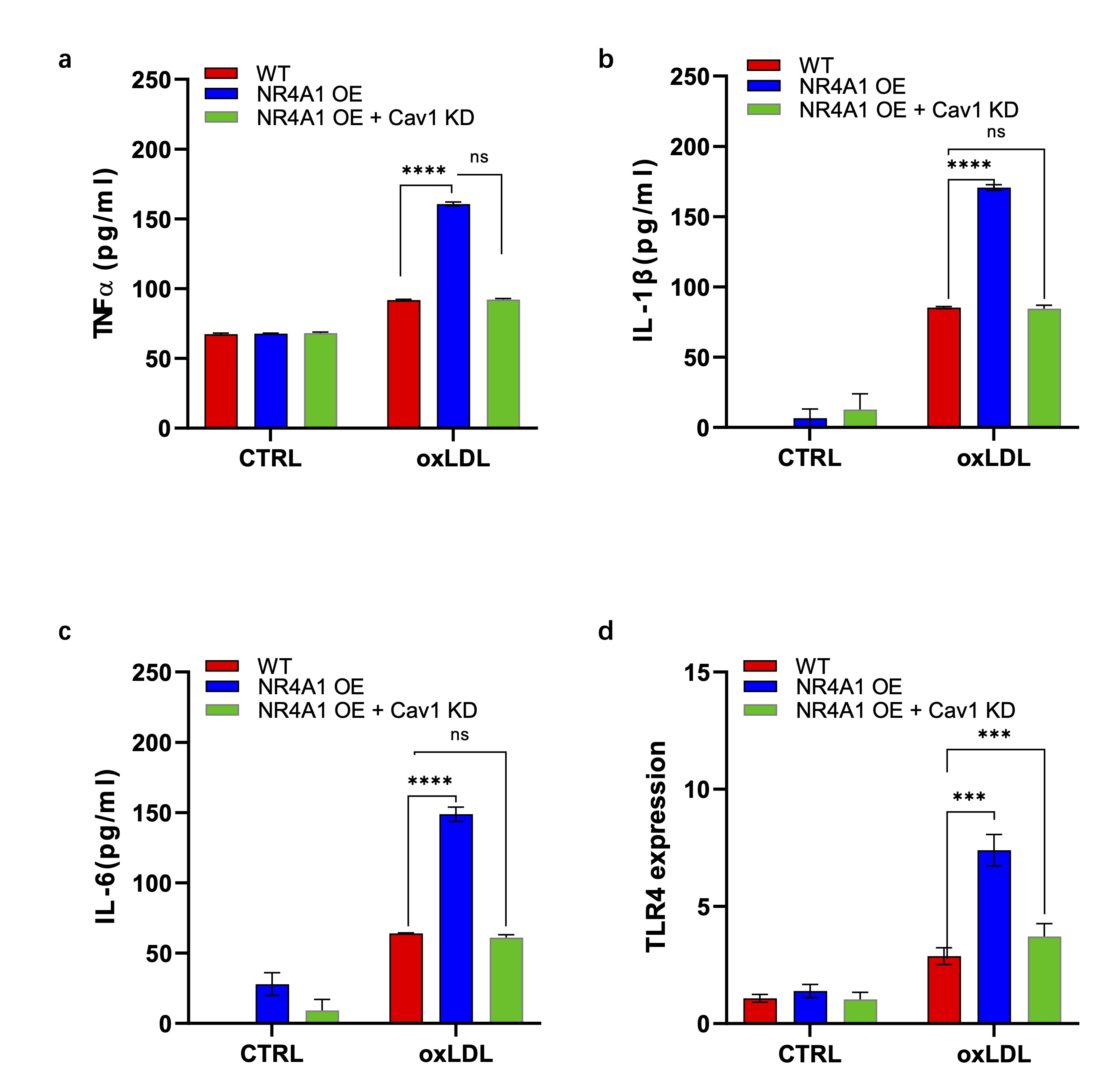
Fig. 6: NR4A1-Caveolin-1 signaling axis promotes inflammatory cytokine expression in senescent macrophage. a-c). Elisa analysis of proinflammatory cytokines TNF- (a), IL-6(b) and IL-1(c) secretions in the supernatants of wild type, NR4A1 overexpression, and NR4A1 overexpression plus caveolin-1 knocking-down Raw264.7 cells with or without oxLDL stimulation. n=3. d). Real-time PCR analysis of proinflammatory gene TLR4 expression in wild type, NR4A1 overexpression, and NR4A1 overexpression plus caveolin-1 knock down Raw264.7 cells with or without oxLDL stimulation. n=3.
Discussion
Oxidative stress is a primary mechanism in the acceleration of aging [39]. Prolonged oxidative stress can induce premature cellular senescence, leading to a state of heightened inflammatory response [40]. Mitochondria and NADPH are the main sources of reactive oxygen species (ROS) in vascular cells [41]. Previous studies have found that ROS produced via NADPH can induce the senescence of macrophage [42]. Interestingly, it has been found that caveolin-1 participates in ROS generation from NADPH, thus affecting the process of macrophage senescence [11]. Meanwhile, mitochondria also play a crucial role in the regulation of senescence. Dysfunction of mitochondria leads to excessive accumulation of ROS, significantly increasing lipid membrane oxidation, disrupting protein synthesis, inducing DNA damage, and promoting premature cell senescence [43]. Activation of autophagy is a potential protective mechanism in the early stage of cellular senescence, promoting cell growth and survival [44]. Oxidative stress enhances mitochondrial autophagy, which, in turn, can regulate antioxidant levels and maintain cellular homeostasis [45]. However, excessive autophagy can exacerbate senescence and cell death [46]. Previous studies have shown that defects in mitochondrial autophagy may increase ROS generation from NADPH [47], while inhibition of ROS derived from NADPH can downregulate cellular mitochondrial autophagy levels [48]. Nevertheless, the regulatory mechanisms of mitochondrial autophagy during the aging process are not yet clear. Our previous study found that nuclear receptor subfamily 4A1 (NR4A1) is an important nuclear transcription factor regulating mitochondrial redox reactions [18]. In the present work, we found that caveolin-1 can promote mitochondrial autophagy during oxLDL-induced cell aging, and this process is further regulated by NR4A1. This discovery provides novel insights into the mechanisms of mitochondrial autophagy and cellular aging. As a nuclear transcription factor, NR4A1 can potentially upregulate caveolin-1 expression through direct transcriptional regulation by binding to promoter regions of the caveolin-1 gene. This possibility is supported by the observed increase in caveolin-1 mRNA and protein levels following NR4A1 overexpression. However, indirect mechanisms—such as NR4A1-mediated signaling cascades or epigenetic modifications—may also contribute to this regulation, and further studies are required to dissect these pathways in detail.
Signaling cascades of autophagy and senescence are tightly correlated, while the specific mechanism regulating autophagy and cellular senescence are not yet clear. Mammalian target of rapamycin (mTOR) is the initiator of autophagy, which is further regulated by several pathways including adenosine monophosphate activated protein kinase (AMPK)/mTOR, phosphatidylinositol 3-kinase (PI3K)/serine-threonine kinase (Akt)/mTOR, and AMPK/silent information regulation 1 (Sirt1) [49-52]. In the context of mitochondrial autophagy or mitophagy, it is regulated by critical elements including PINK1/Parkin, BNIP/NIX, and FUNDC1 [53-55]. Consequently, outer mitochondrial membrane proteins directly or indirectly interact with microtubule-associated protein 1A/1B-light chain 3 (LC3) and p62 to initiate mitophagy and selectively remove damaged mitochondria [56]. In summary, mitophagy is a double-edged sword in regulating oxidative stress-induced premature cellular senescence. Moderate activation of mitophagy has anti-senescence effect, while excessive mitophagy can accelerate cellular senescence or even cell death [57]. Therefore, how to modulate the degree of mitophagy activation is of great clinical significance for treating aging-related diseases.
Mitophagy is closely related to the occurrence and development of AS [58-60]. In AS lesions, endothelial cells, macrophages, and smooth muscle cells exhibit abnormalities in mitophagy [61]. Current research suggests that mitophagy has a dual role in the pathogenesis of AS: basal or moderate mitophagy is an important protective measure for plaque cells against inflammation and oxidative stress reactions [62]. However, excessive cellular autophagy leads to the release of large amounts of inflammatory factors and reactive oxygen species, exacerbating inflammation and oxidative stress reactions [63]. Previous study found that in mice with macrophage-specific deletion of autophagy-related gene ATG5, high-fat diet treatment elevated cell apoptosis, local inflammation, oxidative stress, and necrosis in AS plaques [64]. Basal autophagy of macrophages protects plaque macrophages from various stresses; however, in the late stage of AS, macrophage autophagy is abnormal, leading to changes in macrophage activity, promoting the release of inflammatory factors within the plaque, and thus promoting the progression of AS [65]. It suggested that clearance of macrophage-derived foam cells may delay the progression of AS, while the underlying mechanism, in particular the involvement of mitophagy, has not been defined yet [66].
While the findings presented in this study provide insights into the role of NR4A1 and caveolin-1 in regulating mitochondrial dysfunction during macrophage senescence, several limitations should be considered. First, our study primarily relies on in vitro models, specifically Raw264.7 macrophage cell lines and murine bone marrow-derived macrophages (BMDMs), which may not fully recapitulate the complexity of macrophage behavior in vivo [67]. Although BMDMs provide a more physiologically relevant model, they are still subject to the limitations of in vitro culture conditions and difficulties in genetic manipulations, which may not reflect the systemic and microenvironmental factors present in living organisms. Future studies should aim to validate these findings in vivo using macrophage specific gene overexpression or deletion mouse models, to better understand the physiological relevance of NR4A1 and caveolin-1 in regulating macrophage functions during aging and disease progression. Additionally, while the study highlights the interaction between NR4A1 and caveolin-1 in mitochondrial dysfunction and inflammation, the underlying molecular mechanisms linking these two factors remain incompletely defined. Our findings indicate that NR4A1 enhances the expression of caveolin-1 at both the transcriptional and protein levels during oxLDL-induced senescence. Given that NR4A1 functions as a nuclear receptor, it is plausible that it directly activates caveolin-1 gene transcription. This may occur via binding to response elements in the caveolin-1 promoter region, although indirect regulation through secondary signaling molecules cannot be ruled out. Chromatin immunoprecipitation (ChIP) or promoter reporter assays would be valuable in confirming this direct regulatory relationship.Further research is needed to investigate the detailed signaling pathways that mediate the NR4A1-caveolin-1 axis, particularly focusing on how caveolin-1 modulates mitophagy and mitochondrial integrity [68, 69] . Additionally, the role of other potential regulators of NR4A1 and caveolin-1 expression, such as transcription factors or post-translational modifications, should be explored [70].
Conclusion
In summary, this study elucidates the crucial role of NR4A1 and caveolin-1 in regulating mitophagy during oxLDL-induced macrophage senescence, highlighting a novel regulatory axis that influences cellular aging processes implicated in AS. The findings reveal that NR4A1 plays a pivotal role in oxLDL-induced mitophagy through caveolin-1, and that caveolin-1 itself regulates the expression of NR4A1 under stimulation. This NR4A1-caveolin-1 signaling pathway is essential for the expression of pro-inflammatory cytokines in senescent macrophages, thereby impacting the stability of atherosclerotic plaques.
Acknowledgements
P.L conceived the experiments. P.L, and T.T. performed the experiments and analyzed the data. P.L wrote the manuscript. T.T. and X.H. reviewed and edited the manuscript. All authors read and approved the final manuscript.
This study is supported by Beijing Municipal Natural Science Foundation (Grant Number: 7222070)
Disclosure Statement
The authors have no conflicts of interest to declare.
References
| 1 | Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, Barengo NC, Beaton AZ,
Benjamin EJ, Benziger CP, Bonny A, Brauer M, Brodmann M, Cahill TJ, Carapetis J, Catapano AL, Chugh
SS, Cooper LT, Coresh J, Criqui M, DeCleene N, Eagle KA, Emmons-Bell S, Feigin VL, Fernández-Solà J,
Fowkes G, Gakidou E, Grundy SM, He FJ, Howard G, Hu F, Inker L, Karthikeyan G, Kassebaum N,
Ko/roshetz W, Lavie C, Lloyd-Jones D, Lu HS, Mirijello A, Temesgen AM, Mokdad A, Moran AE, Muntner
P, Narula J, Neal B, Ntsekhe M, Moraes de Oliveira G, Otto C, Owolabi M, Pratt M, Rajagopalan S,
Reitsma M, Ribeiro ALP, Rigotti N, Rodgers A, Sable C, Shakil S, Sliwa-Hahnle K, Stark B, Sundström
J, Timpel P, Tleyjeh IM, Valgimigli M, Vos T, Whelton PK, Yacoub M, Zuhlke L, Murray C, Fuster V:
Global Burden of Cardiovascular Diseases and Risk Factors, 1990-2019: Update From the GBD 2019
Study. J Am Coll Cardiol 2020;76:2982-3021.
https://doi.org/10.1016/j.jacc.2020.11.010 |
| 2 | Tyrrell DJ, Goldstein DR: Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and
potential role of IL-6. Nat Rev Cardiol 2021;18:58-68.
https://doi.org/10.1038/s41569-020-0431-7 |
| 3 | Lee YI, Choi S, Roh WS, Lee JH, Kim TG: Cellular Senescence and Inflammaging in the Skin
Microenvironment. Int J Mol Sci 2021;22
https://doi.org/10.3390/ijms22083849 |
| 4 | Maldonado E, Morales-Pison S, Urbina F, Solari A: Aging Hallmarks and the Role of Oxidative
Stress. Antioxidants 2023;12:651.
https://doi.org/10.3390/antiox12030651 |
| 5 | Guo J, Huang X, Dou L, Yan M, Shen T, Tang W, Li J: Aging and aging-related diseases: from
molecular mechanisms to interventions and treatments. Signal Transduction and Targeted Therapy
2022;7:391.
https://doi.org/10.1038/s41392-022-01251-0 |
| 6 | Hu C, Zhang X, Teng T, Ma ZG, Tang QZ: Cellular Senescence in Cardiovascular Diseases: A
Systematic Review. Aging Dis 2022;13:103-128.
https://doi.org/10.14336/AD.2021.0927 |
| 7 | Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO: Cellular senescence: the good, the bad and
the unknown. Nat Rev Nephrol 2022;18:611-627.
https://doi.org/10.1038/s41581-022-00601-z |
| 8 | Vellasamy DM, Lee SJ, Goh KW, Goh BH, Tang YQ, Ming LC, Yap WH: Targeting Immune Senescence in
Atherosclerosis. Int J Mol Sci 2022;23
https://doi.org/10.3390/ijms232113059 |
| 9 | Wang JC, Bennett M: Aging and atherosclerosis: mechanisms, functional consequences, and potential
therapeutics for cellular senescence. Circ Res 2012;111:245-259.
https://doi.org/10.1161/CIRCRESAHA.111.261388 |
| 10 | Grufman H, Schiopu A, Edsfeldt A, Björkbacka H, Nitulescu M, Nilsson M, Persson A, Nilsson J,
Gonçalves I: Evidence for altered inflammatory and repair responses in symptomatic carotid plaques
from elderly patients. Atherosclerosis 2014;237:177-182.
https://doi.org/10.1016/j.atherosclerosis.2014.08.042 |
| 11 | Wang J, Bai Y, Zhao X, Ru J, Kang N, Tian T, Tang L, An Y, Li P: oxLDL-mediated cellular
senescence is associated with increased NADPH oxidase p47phox recruitment to caveolae. Biosci Rep
2018;38
https://doi.org/10.1042/BSR20180283 |
| 12 | Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, Jang MY, Seok Jang H, Yun TJ, Lee SH, Yoon
WK, Prat A, Seidah NG, Choi J, Lee SP, Yoon SH, Nam JW, Seong JK, Oh GT, Randolph GJ, Artyomov MN,
Cheong C, Choi JH: Transcriptome Analysis Reveals Nonfoamy Rather Than Foamy Plaque Macrophages Are
Proinflammatory in Atherosclerotic Murine Models. Circ Res 2018;123:1127-1142.
https://doi.org/10.1161/CIRCRESAHA.118.312804 |
| 13 | Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV: Mitochondrial aging and age-related
dysfunction of mitochondria. Biomed Res Int 2014;2014:238463.
https://doi.org/10.1155/2014/238463 |
| 14 | Tanaka A: Parkin-mediated selective mitochondrial autophagy, mitophagy: Parkin purges damaged
organelles from the vital mitochondrial network. FEBS Lett 2010;584:1386-1392.
https://doi.org/10.1016/j.febslet.2010.02.060 |
| 15 | Choi SH, Agatisa-Boyle C, Gonen A, Kim A, Kim J, Alekseeva E, Tsimikas S, Miller YI: Intracellular
AIBP (Apolipoprotein A-I Binding Protein) Regulates Oxidized LDL (Low-Density Lipoprotein)-Induced
Mitophagy in Macrophages. Arterioscler Thromb Vasc Biol 2021;41:e82-e96.
https://doi.org/10.1161/ATVBAHA.120.315485 |
| 16 | Tai H, Wang Z, Gong H, Han X, Zhou J, Wang X, Wei X, Ding Y, Huang N, Qin J, Zhang J, Wang S, Gao
F, Chrzanowska-Lightowlers ZM, Xiang R, Xiao H: Autophagy impairment with lysosomal and
mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence.
Autophagy 2017;13:99-113.
https://doi.org/10.1080/15548627.2016.1247143 |
| 17 | Cheng Z, Völkers M, Din S, Avitabile D, Khan M, Gude N, Mohsin S, Bo T, Truffa S, Alvarez R, Mason
M, Fischer KM, Konstandin MH, Zhang XK, Heller Brown J, Sussman MA: Mitochondrial translocation of
Nur77 mediates cardiomyocyte apoptosis. Eur Heart J 2011;32:2179-2188.
https://doi.org/10.1093/eurheartj/ehq496 |
| 18 | Li P, Bai Y, Zhao X, Tian T, Tang L, Ru J, An Y, Wang J: NR4A1 contributes to high-fat associated
endothelial dysfunction by promoting CaMKII-Parkin-mitophagy pathways. Cell Stress Chaperones
2018;23:749-761.
https://doi.org/10.1007/s12192-018-0886-1 |
| 19 | Hanna RN, Carlin LM, Hubbeling HG, Nackiewicz D, Green AM, Punt JA, Geissmann F, Hedrick CC: The
transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C-
monocytes. Nat Immunol 2011;12:778-785.
https://doi.org/10.1038/ni.2063 |
| 20 | Bonta PI, van Tiel CM, Vos M, Pols TW, van Thienen JV, Ferreira V, Arkenbout EK, Seppen J, Spek
CA, van der Poll T, Pannekoek H, de Vries CJ: Nuclear receptors Nur77, Nurr1, and NOR-1 expressed in
atherosclerotic lesion macrophages reduce lipid loading and inflammatory responses. Arterioscler
Thromb Vasc Biol 2006;26:2288-2294.
https://doi.org/10.1161/01.ATV.0000238346.84458.5d |
| 21 | Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, Zaugg C, Pei H, Geissmann F, Ley K,
Hedrick CC: NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and
increases atherosclerosis. Circ Res 2012;110:416-427.
https://doi.org/10.1161/CIRCRESAHA.111.253377 |
| 22 | Nus M, Basatemur G, Galan M, Cros-Brunsó L, Zhao TX, Masters L, Harrison J, Figg N, Tsiantoulas D,
Geissmann F, Binder CJ, Sage AP, Mallat Z: NR4A1 Deletion in Marginal Zone B Cells Exacerbates
Atherosclerosis in Mice-Brief Report. Arterioscler Thromb Vasc Biol 2020;40:2598-2604.
https://doi.org/10.1161/ATVBAHA.120.314607 |
| 23 | Filippini A, D'Alessio A: Caveolae and Lipid Rafts in Endothelium: Valuable Organelles for
Multiple Functions. Biomolecules 2020;10
https://doi.org/10.3390/biom10091218 |
| 24 | Kenworthy AK: The building blocks of caveolae revealed: caveolins finally take center stage.
Biochem Soc Trans 2023;51:855-869.
https://doi.org/10.1042/BST20221298 |
| 25 | Zou H, Stoppani E, Volonte D, Galbiati F: Caveolin-1, cellular senescence and age-related
diseases. Mech Ageing Dev 2011;132:533-542.
https://doi.org/10.1016/j.mad.2011.11.001 |
| 26 | Volonte D, Zhang K, Lisanti MP, Galbiati F: Expression of caveolin-1 induces premature cellular
senescence in primary cultures of murine fibroblasts. Mol Biol Cell 2002;13:2502-2517.
https://doi.org/10.1091/mbc.01-11-0529 |
| 27 | Yu DM, Jung SH, An HT, Lee S, Hong J, Park JS, Lee H, Lee H, Bahn MS, Lee HC, Han NK, Ko J, Lee
JS, Ko YG: Caveolin-1 deficiency induces premature senescence with mitochondrial dysfunction. Aging
Cell 2017;16:773-784.
https://doi.org/10.1111/acel.12606 |
| 28 | Volonte D, Liu Z, Shiva S, Galbiati F: Caveolin-1 controls mitochondrial function through
regulation of m-AAA mitochondrial protease. Aging (Albany NY) 2016;8:2355-2369.
https://doi.org/10.18632/aging.101051 |
| 29 | Hou K, Li S, Zhang M, Qin X: Caveolin-1 in autophagy: A potential therapeutic target in
atherosclerosis. Clinica Chimica Acta 2021;513:25-33.
https://doi.org/10.1016/j.cca.2020.11.020 |
| 30 | Jiang Y, Krantz S, Qin X, Li S, Gunasekara H, Kim YM, Zimnicka A, Bae M, Ma K, Toth PT, Hu Y,
Shajahan-Haq AN, Patel HH, Gentile S, Bonini MG, Rehman J, Liu Y, Minshall RD: Caveolin-1 controls
mitochondrial damage and ROS production by regulating fission - fusion dynamics and mitophagy. Redox
Biol 2022;52:102304.
https://doi.org/10.1016/j.redox.2022.102304 |
| 31 | Iggena D, Winter Y, Steiner B: Melatonin restores hippocampal neural precursor cell proliferation
and prevents cognitive deficits induced by jet lag simulation in adult mice. J Pineal Res 2017;62
https://doi.org/10.1111/jpi.12397 |
| 32 | Toda G, Yamauchi T, Kadowaki T, Ueki K: Preparation and culture of bone marrow-derived macrophages
from mice for functional analysis. STAR Protoc 2021;2:100246.
https://doi.org/10.1016/j.xpro.2020.100246 |
| 33 | Hu SY, Zhang Y, Zhu PJ, Zhou H, Chen YD: Liraglutide directly protects cardiomyocytes against
reperfusion injury possibly via modulation of intracellular calcium homeostasis. J Geriatr Cardiol
2017;14:57-66.
|
| 34 | Zhou H, Li D, Shi C, Xin T, Yang J, Zhou Y, Hu S, Tian F, Wang J, Chen Y: Effects of Exendin-4 on
bone marrow mesenchymal stem cell proliferation, migration and apoptosis in vitro. Scientific
Reports 2015;5:12898.
https://doi.org/10.1038/srep12898 |
| 35 | Huang J-r, Zhang M-h, Chen Y-j, Sun Y-l, Gao Z-m, Li Z-j, Zhang G-p, Qin Y, Dai X-y, Yu X-y, Wu
X-q: Urolithin A ameliorates obesity-induced metabolic cardiomyopathy in mice via mitophagy
activation. Acta Pharmacologica Sinica 2023;44:321-331.
https://doi.org/10.1038/s41401-022-00919-1 |
| 36 | Sheng J, Li H, Dai Q, Lu C, Xu M, Zhang J, Feng J: NR4A1 Promotes Diabetic Nephropathy by
Activating Mff-Mediated Mitochondrial Fission and Suppressing Parkin-Mediated Mitophagy. Cell
Physiol Biochem 2018;48:1675-1693.
https://doi.org/10.1159/000492292 |
| 37 | Oishi Y, Manabe I: Macrophages in age-related chronic inflammatory diseases. npj Aging and
Mechanisms of Disease 2016;2:16018.
https://doi.org/10.1038/npjamd.2016.18 |
| 38 | Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL: Cellular senescence and the senescent
secretory phenotype: therapeutic opportunities. J Clin Invest 2013;123:966-972.
https://doi.org/10.1172/JCI64098 |
| 39 | Tyrrell DJ, Blin MG, Song J, Wood SC, Zhang M, Beard DA, Goldstein DR: Age-Associated
Mitochondrial Dysfunction Accelerates Atherogenesis. Circ Res 2020;126:298-314.
https://doi.org/10.1161/CIRCRESAHA.119.315644 |
| 40 | Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, Gargiulo G, Testa G, Cacciatore F,
Bonaduce D, Abete P: Oxidative stress, aging, and diseases. Clin Interv Aging 2018;13:757-772.
https://doi.org/10.2147/CIA.S158513 |
| 41 | Dan Dunn J, Alvarez LAJ, Zhang X, Soldati T: Reactive oxygen species and mitochondria: A nexus of
cellular homeostasis. Redox Biology 2015;6:472-485.
https://doi.org/10.1016/j.redox.2015.09.005 |
| 42 | Li H, Luo YF, Wang YS, Yang Q, Xiao YL, Cai HR, Xie CM: Using ROS as a Second Messenger, NADPH
Oxidase 2 Mediates Macrophage Senescence via Interaction with NF-κB during Pseudomonas aeruginosa
Infection. Oxid Med Cell Longev 2018;2018:9741838.
https://doi.org/10.1155/2018/9741838 |
| 43 | Guo C, Sun L, Chen X, Zhang D: Oxidative stress, mitochondrial damage and neurodegenerative
diseases. Neural Regen Res 2013;8:2003-2014.
|
| 44 | Cassidy LD, Narita M: Autophagy at the intersection of aging, senescence, and cancer. Mol Oncol
2022;16:3259-3275.
https://doi.org/10.1002/1878-0261.13269 |
| 45 | Lee J, Giordano S, Zhang J: Autophagy, mitochondria and oxidative stress: cross-talk and redox
signalling. Biochemical Journal 2011;441:523-540.
https://doi.org/10.1042/BJ20111451 |
| 46 | Kwon Y, Kim JW, Jeoung JA, Kim MS, Kang C: Autophagy Is Pro-Senescence When Seen in Close-Up, but
Anti-Senescence in Long-Shot. Mol Cells 2017;40:607-612.
https://doi.org/10.14348/molcells.2017.0151 |
| 47 | Sciarretta S, Zhai P, Shao D, Zablocki D, Nagarajan N, Terada LS, Volpe M, Sadoshima J: Activation
of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during
energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic
initiation factor 2α/activating transcription factor 4 pathway. Circ Res 2013;113:1253-1264.
https://doi.org/10.1161/CIRCRESAHA.113.301787 |
| 48 | Choi SH, Gonen A, Diehl CJ, Kim J, Almazan F, Witztum JL, Miller YI: SYK regulates macrophage
MHC-II expression via activation of autophagy in response to oxidized LDL. Autophagy
2015;11:785-795.
https://doi.org/10.1080/15548627.2015.1037061 |
| 49 | Panwar V, Singh A, Bhatt M, Tonk RK, Azizov S, Raza AS, Sengupta S, Kumar D, Garg M: Multifaceted
role of mTOR (mammalian target of rapamycin) signaling pathway in human health and disease. Signal
Transduct Target Ther 2023;8:375.
https://doi.org/10.1038/s41392-023-01608-z |
| 50 | Kim J, Kundu M, Viollet B, Guan KL: AMPK and mTOR regulate autophagy through direct
phosphorylation of Ulk1. Nat Cell Biol 2011;13:132-141.
https://doi.org/10.1038/ncb2152 |
| 51 | Ferreira-Marques M, Carvalho A, Cavadas C, Aveleira CA: PI3K/AKT/MTOR and ERK1/2-MAPK signaling
pathways are involved in autophagy stimulation induced by caloric restriction or caloric restriction
mimetics in cortical neurons. Aging (Albany NY) 2021;13:7872-7882.
https://doi.org/10.18632/aging.202805 |
| 52 | Siddhi J, Sherkhane B, Kalavala AK, Arruri V, Velayutham R, Kumar A: Melatonin prevents
diabetes-induced nephropathy by modulating the AMPK/SIRT1 axis: Focus on autophagy and mitochondrial
dysfunction. Cell Biology International 2022;46:2142-2157.
https://doi.org/10.1002/cbin.11899 |
| 53 | Whitworth AJ, Pallanck LJ: PINK1/Parkin mitophagy and neurodegeneration-what do we really know in
vivo? Current Opinion in Genetics & Development 2017;44:47-53.
https://doi.org/10.1016/j.gde.2017.01.016 |
| 54 | Choi GE, Lee HJ, Chae CW, Cho JH, Jung YH, Kim JS, Kim SY, Lim JR, Han HJ: BNIP3L/NIX-mediated
mitophagy protects against glucocorticoid-induced synapse defects. Nat Commun 2021;12:487.
https://doi.org/10.1038/s41467-020-20679-y |
| 55 | Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B,
Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q: Mitochondrial outer-membrane protein
FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nature Cell Biology 2012;14:177-185.
https://doi.org/10.1038/ncb2422 |
| 56 | Yildirim RM, Ergun Y, Basar M: Mitochondrial Dysfunction, Mitophagy and Their Correlation with
Perinatal Complications: Preeclampsia and Low Birth Weight. Biomedicines 2022;10
https://doi.org/10.3390/biomedicines10102539 |
| 57 | Miwa S, Kashyap S, Chini E, von Zglinicki T: Mitochondrial dysfunction in cell senescence and
aging. J Clin Invest 2022;132
https://doi.org/10.1172/JCI158447 |
| 58 | Poznyak AV, Nikiforov NG, Wu W-K, Kirichenko TV, Orekhov AN: Autophagy and Mitophagy as Essential
Components of Atherosclerosis. Cells 2021;10:443.
https://doi.org/10.3390/cells10020443 |
| 59 | Orekhov AN, Poznyak AV, Sobenin IA, Nikifirov NN, Ivanova EA: Mitochondrion as a Selective Target
for the Treatment of Atherosclerosis: Role of Mitochondrial DNA Mutations and Defective Mitophagy in
the Pathogenesis of Atherosclerosis and Chronic Inflammation. Curr Neuropharmacol 2020;18:1064-1075.
https://doi.org/10.2174/1570159X17666191118125018 |
| 60 | Ma S, Chen J, Feng J, Zhang R, Fan M, Han D, Li X, Li C, Ren J, Wang Y, Cao F: Melatonin
Ameliorates the Progression of Atherosclerosis via Mitophagy Activation and NLRP3 Inflammasome
Inhibition. Oxidative Medicine and Cellular Longevity 2018;2018:9286458.
https://doi.org/10.1155/2018/9286458 |
| 61 | Poznyak AV, Nikiforov NG, Wu WK, Kirichenko TV, Orekhov AN: Autophagy and Mitophagy as Essential
Components of Atherosclerosis. Cells 2021;10
https://doi.org/10.3390/cells10020443 |
| 62 | Swiader A, Nahapetyan H, Faccini J, D'Angelo R, Mucher E, Elbaz M, Boya P, Vindis C: Mitophagy
acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by
atherogenic lipids. Oncotarget 2016;7:28821-28835.
https://doi.org/10.18632/oncotarget.8936 |
| 63 | Doblado L, Lueck C, Rey C, Samhan-Arias AK, Prieto I, Stacchiotti A, Monsalve M: Mitophagy in
Human Diseases. Int J Mol Sci 2021;22
https://doi.org/10.3390/ijms22083903 |
| 64 | Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL: Autophagy regulates cholesterol efflux
from macrophage foam cells via lysosomal acid lipase. Cell Metab 2011;13:655-667.
https://doi.org/10.1016/j.cmet.2011.03.023 |
| 65 | Sergin I, Razani B: Self-eating in the plaque: what macrophage autophagy reveals about
atherosclerosis. Trends Endocrinol Metab 2014;25:225-234.
https://doi.org/10.1016/j.tem.2014.03.010 |
| 66 | Martinet W, Coornaert I, Puylaert P, De Meyer GRY: Macrophage Death as a Pharmacological Target in
Atherosclerosis. Front Pharmacol 2019;10:306.
https://doi.org/10.3389/fphar.2019.00306 |
| 67 | Moore KJ, Sheedy FJ, Fisher EA: Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol
2013;13:709-721.
https://doi.org/10.1038/nri3520 |
| 68 | Díaz-Valdivia N, Simón L, Díaz J, Martinez-Meza S, Contreras P, Burgos-Ravanal R, Pérez VI, Frei
B, Leyton L, Quest AFG: Mitochondrial Dysfunction and the Glycolytic Switch Induced by Caveolin-1
Phosphorylation Promote Cancer Cell Migration, Invasion, and Metastasis. Cancers 2022;14:2862.
https://doi.org/10.3390/cancers14122862 |
| 69 | Nah J, Yoo S-M, Jung S, Jeong EI, Park M, Kaang B-K, Jung Y-K: Phosphorylated CAV1 activates
autophagy through an interaction with BECN1 under oxidative stress. Cell Death & Disease
2017;8:e2822-e2822.
https://doi.org/10.1038/cddis.2017.71 |
| 70 | Deng S, Chen B, Huo J, Liu X: Therapeutic potential of NR4A1 in cancer: Focus on metabolism.
Frontiers in Oncology 2022;12.
https://doi.org/10.3389/fonc.2022.972984 |